
Tumor-Suppressor Genes
If the first class of cancer-inducing mutations steps on the accelerator of cell division, the second class of cancer-inducing mutations removes the brakes. Cell division is normally turned off in healthy cells by proteins that prevent cyclins from binding to Cdks. The genes that encode these proteins are called tumor-suppressor genes. Their mutant alleles are genetically recessive.
One of the first tumor-suppressor genes to be discovered was the Rb gene. The role of Rb is to bind to the transcription factor E2F, preventing its activity. The E2F transcription factor is necessary for the expression of a number of cell-cycle-specific genes. In the normal course of the cell cycle, the action of cyclin-dependent kinase acts to phosphorylate Rb. As phosphorylated Rb cannot bind E2F, this releases E2F from its inhibition and allows the cell cycle to progress.
A loss of normal Rb function leads to the loss of control over the cell cycle (figure 5). However, both normally functioning copies of Rb must be lost before control is removed. Thus the cancer-inducing mutant alleles of Rb are genetically recessive.
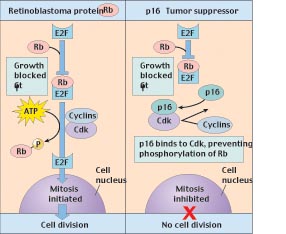
Figure 5
How the tumor-suppressor genes Rb and p16 interact to block cell division. The retinoblastoma protein (Rb) binds to the transcription factor (E2F) that activates genes in the nucleus, preventing this factor from initiating mitosis. The G1 checkpoint is passed when Cdk interacts with cyclins to phosphorylate Rb, releasing E2F. The p16 tumor-suppressor protein reinforces Rbs inhibitory action by binding to Cdk so that Cdk is not available to phosphorylate Rb.
Another well characterized tumor suppressor gene is p53, a gene found to be mutated in a large proportion of human cancers. The role of p53 is to integrate siganls that sense DNA damage during G1 and G2 (figure 6). When significant DNA damage is detected, p53 induces apoptosis to remove the damaged cell. Loss of p53 leads to acumulation of mutations that would have otherwise been removed, and thus failure to prevent transformation to cancerous growth. One of the reasons repeated smoking leads inexorably to lung cancer is that it induces p53 mutations. Indeed, almost half of all cancers involve mutations of the p53 gene.
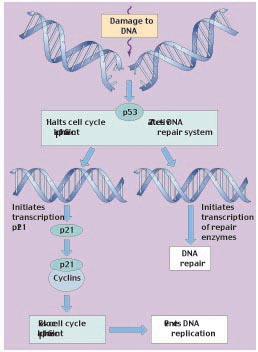
Figure 6
The role of tumor-suppressor p53 in regulating the cell cycle. The p53 protein works at the G1 checkpoint to check for DNA damage. If the DNA is damaged, p53 activates the DNA repair system and stops the cell cycle at the G1 checkpoint (before DNA replication). This allows time for the damage to be repaired. p53 stops the cell cycle by inducing the transcription of p21. The p21 protein then binds to cyclins and prevents them from complexing with Cdk.
Epigenetic Events and Cancer
Recent research on the relationship between chromatin structure and the regulation of gene expression also shed light on the process of malignant transformation. The role of methylation and histone deacetylation has a bearing on cancer as well. The majority of methylation of mammalian DNA is of cytosine at sites where cytosine is followed by guanine in the DNA sequence (called CpG repeat sites). Most of the CpG repeats found in humans are methylated. Those that are not, so-called CpG islands, correlate with genes that are expressed. Recently, hypermethylation of CpG islands has been observed in a number of human cancers. Although it is not yet entirely clear that hypermethylation is a direct cause of cancer, rather than a result of it, the finding that key tumor suppressor genes may exhibit this hypermethylation strengthens the case for this epigenetic process as a contributing cause. And even if the hypermethylation is not the cancer-initiating event, aberrant methylation patterns in cancer cells may shut off additional genes, accelerating the process. This provides another potential mechanism of inactivation of tumor-suppressor genes to add to our growing list: point mutation, structural alterations of chromosomes, and now epigenetic control of gene exdpression.
Cancer Is a Multi-Step Process
Cells control proliferation at several checkpoints, and all of these controls must be inactivated for cancer to be initiated. Therefore, the induction of most cancers involves the mutation of multiple genes; four to six is a typical number (figure 7). In many of the tissue culture cell lines used to study cancer, most of the controls are already inactivated, so that mutations in only one or a few genes transform the line into cancerous growth. The need to inactivate several regulatory genes almost certainly explains why most cancers occur in people over 40 years old (figure 8); in older persons, there has been more time for individual cells to accumulate multiple mutations. It is now clear that mutations, including those in potentially cancer-causing genes, do accumulate over time. Using the polymerase chain reaction (PCR), researchers in 1994 searched for a certain cancer-associated gene mutation in the blood cells of 63 cancer-free people. They found that the mutation occurred 13 times more often in people over 60 years old than in people under 20.
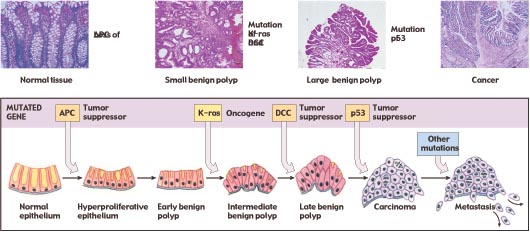
Figure 7
The progression of mutations that commonly lead to colorectal cancer. The fatal metastasis is the last of six serial changes that the epithelial cells lining the rectum undergo. One of these changes is brought about by mutation of a proto-oncogene, and three of them involve mutations that inactivate tumor-suppressor genes.
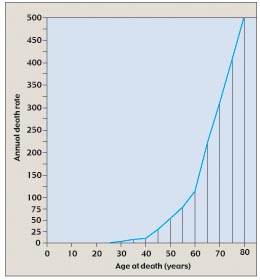
Figure 8
The annual death rate from cancer climbs with age. The rate of cancer deaths increases steeply after age 40 and even more steeply after age 60, suggesting that several independent mutations must accumulate to give rise to cancer.
Cancer is a disease in which the controls that normally restrict cell proliferation do not operate. In some cases, cancerous growth is initiated by the inappropriate activation of proteins that regulate the cell cycle; in other cases, it is initiated by the inactivation of proteins that normally suppress cell division.