
Human Genome Project
Variation Within Genome
One of the most surprising results revealed by genome sequences has been the discovery of just how similar living things are to one another at the genetic level. Forty-two percent of the genes discovered in C. elegans had some sort of match to genes in other organisms only distantly related. Fully 83% of Drosophila genes match those of other species. The matches are not perfect howeverthe DNA sequences of genes that do the same job have drifted apart over millions of years. But functionality is maintained. For example, when a gene involved in eye development in mice was substituted for its homologue in Drosophila, the flies were born with normal, functional eyes.
Perhaps the most striking lesson learned from the sequence of the human genome is how very like other organisms humans are. More than half of the genes of Drosophila have human counterparts. Among mammals the differences are even fewer. Humans have only 300 genes that have no counterpart in the mouse genome. This suggests that when an ape genome is sequenced, it will possess practically all of the genes that humans do.
Single Nucleotide Polymorphisms (SNPs)
While the sequence of the human genome is readily distinguishable from other species, there is tremendous variation among individual human genomes. So far we have just two genomes belonging to anonymous individuals. The private company used a male with the code name Celer (a first century architect who broke with Roman tradition and created a new architectural form). Twelve men and women contributed DNA to Mosaic Mans sequence, the public genome. However, most of the sequence was based on a single male.
SNPs (pronounced snips) appear to be the most effective way to analyze variation at the whole genome level. SNPs, or single nucleotide polymorphisms, are variations (polymorphisms) in single base pairs (figure 10.4). For example, you might have the sequence ACGCTCA, while a friend has the sequence AGGCTCA in the same gene. Two of every three SNPs are the result of replacement of C with T. About 1.42 million SNPs are distributed across our genome. That is, about one every 1200 bases that are mapped onto the human genome are SNPs. SNPs can serve as markers of particular human alleles, such as those that cause inherited diseases. About 60,000 SNPs have been identified in exons, and 85% of all exons are less than 5 kb from a identified SNP.
SNPs have a low rate of recurrent mutation, so they are excellent markers of human history. Iceland provides a clear example. Almost no immigration occurred in Iceland for more than a thousand years, so the gene pool among Icelanders today reflects Northern Europe about 800 A.D. The BRCA2 breast cancer gene in the Iceland has only one mutant allele, dating back to a mutation in a sixteenth century cleric named Einar. A collective genetic and genealogical database being created in Iceland will provide a unique opportunity to study the evolution of SNPs within the human genome.
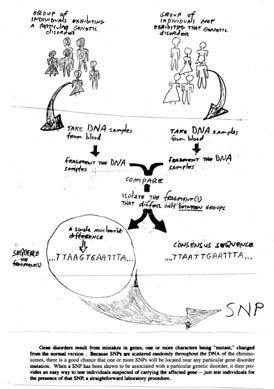
Figure 10.4 Using SNPs to screen for genetic disorders.
Many genetic disorders result from point mutations in genes. Because SNPs are scattered randomly throughout the genome, there is a good chance that one or more SNPs will be located near any particular mutation. Once a SNP has been shown to be associated with a mutation causing a genetic disorder, it can be used as an indicator of the diseasejust test individuals for the presence of that SNP.
Genomic Exchanges Between Species
Comparisons of the genomes of different organisms suggests that genes have also moved laterally between organisms. For example, humans have several hundred genes similar to those of bacteria. Because these genes have no counterpoint in roundworms and fruit flies, researchers surmise the genes were acquired from bacteria by lateral transfer. An example is the gene encoding monoamine oxidase, an important degradative enzyme for the central nervous system.
The many transposons of the human genome also provide a paleontological record of the human genome over several hundred million years. Comparisons among versions of a transposon that has duplicated many times allow researchers to construct a family tree to identify the ancestral form of the transposon. The percent sequence divergence of the duplicates allows the researcher to estimate the time when that particular transposon originally invaded the human genome millions of years ago.